
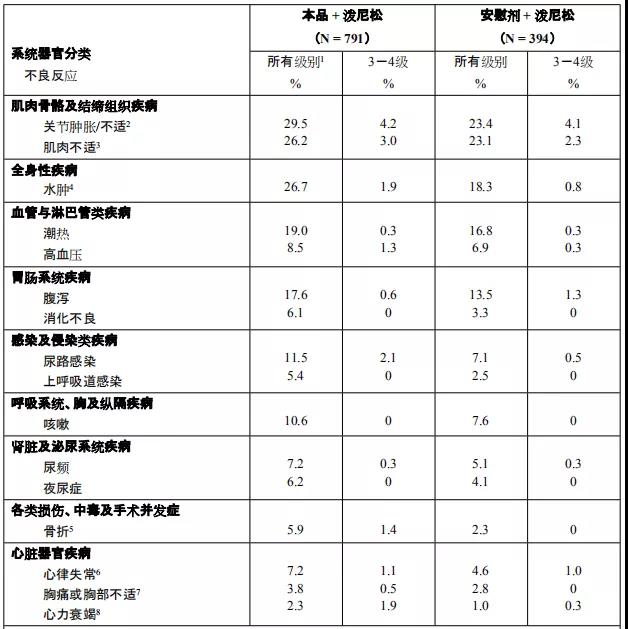
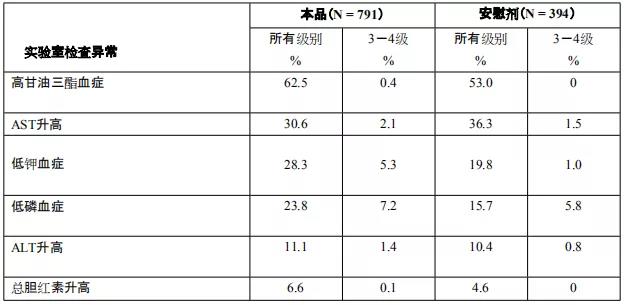
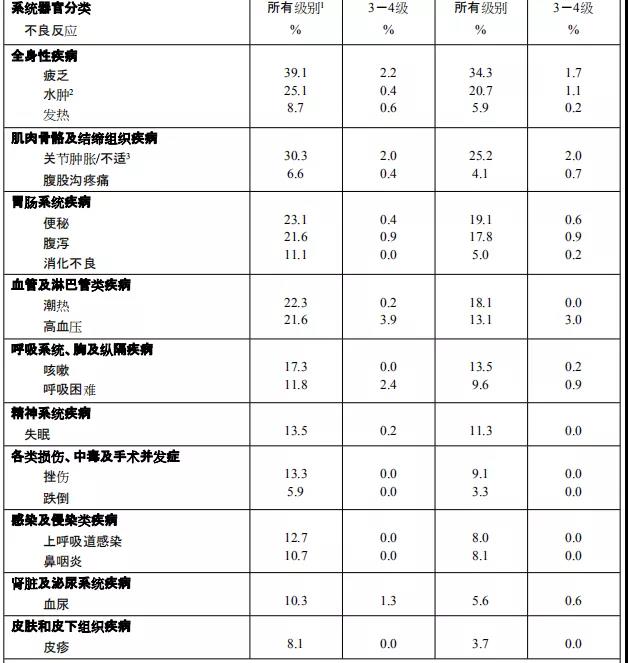

阿比特龙是由强生公司研制开发,为口服有效的CYP17A酶不可逆抑制剂, 2011年4月获得美国FDA批准,商品名为“Zytiga”。主要对睾丸、前列腺和肾上腺组织中的雄性激素均有抑制作用。
该产品凭借其可使前列腺癌患者体内前列腺特异性抗原水平显著下降,利于肿瘤萎缩,使前列腺癌晚期患者的寿命延长数年等特点,阿比特龙上市不久便得到认可,全球销售额增长迅速,很快便成为强生公司的重磅产品之一。
强生的阿比特龙于2015年5月在我国CFDA获得批准上市,商品名为“泽珂”,与泼尼松联用治疗转移性去势抵抗性前列腺癌(mCRPC)患者;2018年12月,阿比特龙新适应症以优先审评方式在中国获批,与泼尼松或泼尼松龙联用一线治疗新诊断的高危转移性内分泌治疗敏感性前列腺癌(mHSPC),包括未接受过内分泌治疗或接受内分泌治疗≤3个月的mHSPC患者。