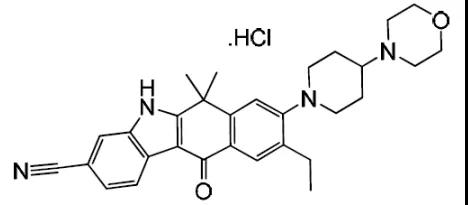
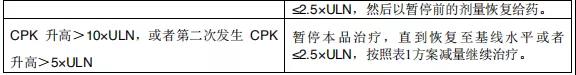
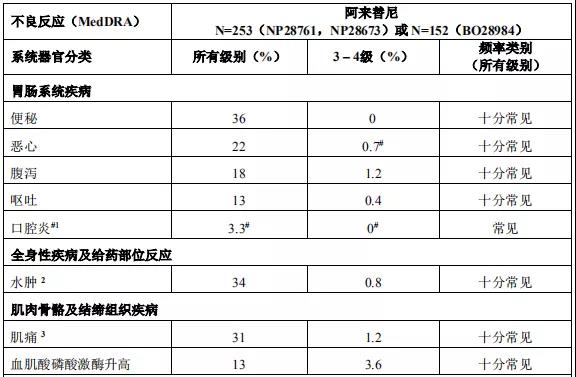
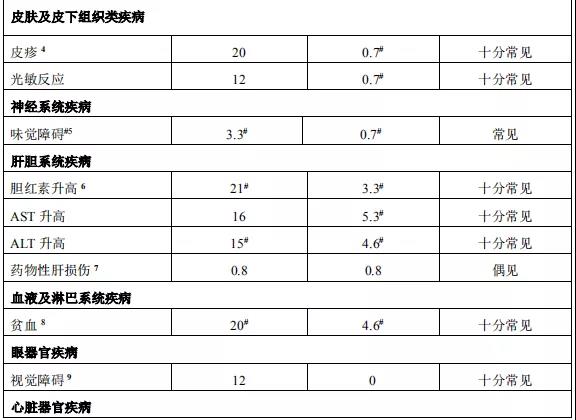
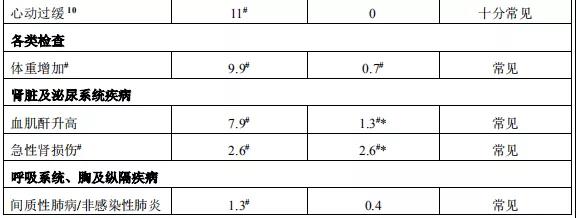

2015年12月11日, 阿来替尼首次获得FDA批准上市 。安圣莎®(阿来替尼)分别于2017年11月和12月在美国与欧盟获批一线治疗ALK阳性非小细胞肺癌。
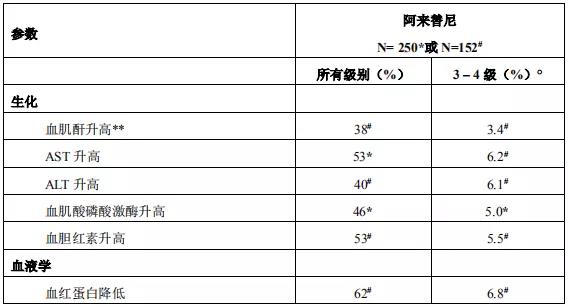
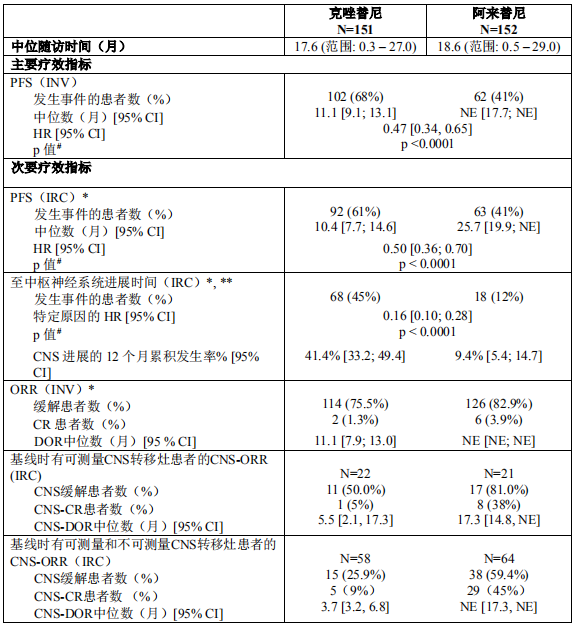
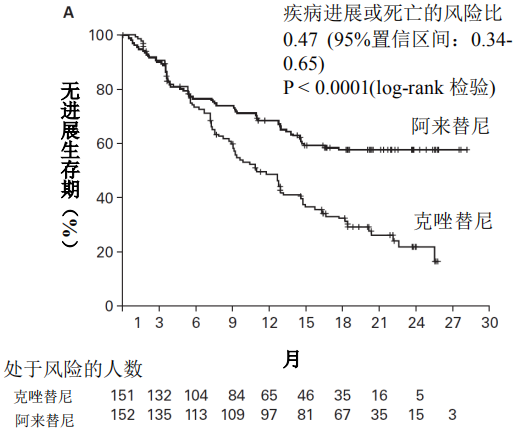
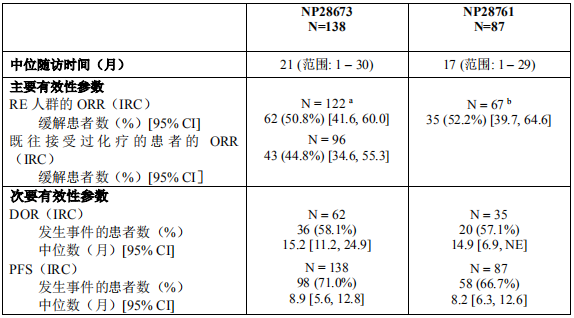
2018年3月进入国家药品监督管理局药品审评中心优先审评程序,8月15日国家药品监督管理局正式批准了罗氏安圣莎(阿来替尼)的进口注册申请,用于治疗间变性淋巴瘤激酶(Anaplastic Lymphoma Kinase,ALK)阳性的局部晚期或转移性非小细胞肺癌(NSCLC)。阿来替尼目前是Best in class的ALK抑制剂,为中国ALK阳性非小细胞肺癌患者提供了新的治疗选择。
2018年3月进入国家药品监督管理局药品审评中心优先审评程序,8月15日国家药品监督管理局正式批准了罗氏安圣莎(阿来替尼)的进口注册申请,用于治疗间变性淋巴瘤激酶(Anaplastic Lymphoma Kinase,ALK)阳性的局部晚期或转移性非小细胞肺癌(NSCLC)。阿来替尼目前是Best in class的ALK抑制剂,为中国ALK阳性非小细胞肺癌患者提供了新的治疗选择。