Science 癌症特刊:针对 RAS 基因的靶向药物研发盘点
近十多年来,人类对于癌症的复杂性有了进一步的了解。不同患者之间、原发或转移性肿瘤之间、甚至是不同区域的同一种肿瘤之间,在遗传上都存在着大量不一致。这种肿瘤的异质性能够解释为何药物的作用因人而异,也能够解释患者为何会出现耐药性的复发。诚然,癌症的复杂性为研究人员们提出了严峻的挑战,但它也不断推动着癌症疗法的进展。近期,《科学》杂志推出了癌症特刊,介绍了目前处于前沿的癌症疗法。今日,我们来了解一下,针对 RAS 基因的靶向药物研发,已经有了怎样的进展。

RAS 是第一个在人类肿瘤中被鉴定出来的致癌基因,时至今日,三个 RAS 基因也是在癌症中最常见的致癌基因——大约 25% 的人类肿瘤里存在 RAS 突变。科学家们早就发现,RAS 基因突变会导致它持续的过度活跃,从而促使癌细胞生长。再加上 RAS 突变的广泛存在,它实在是一个理论上理想的药物靶点。但是,自 RAS 蛋白被发现 30 多年以来,还没有一个针对 RAS 的药物被成功研发出来,这也让一些人认为 RAS 是不可成药的。不过,很多科研人员并没有放弃,2013 年美国国家癌症研究院(National Cancer Institute)专门提出一项倡议,对 RAS“宣战”。最近几年对于 RAS 本身和 RAS 靶向药物研究取得了很大的进展。

▲五种靶向 RAS 蛋白的思路(图片来源:《科学》)
肿瘤中的 RAS 突变
目前已知的 RAS 家族共有三个基因:KRAS,NRAS 和 HRAS。在人类肿瘤中,KRAS 突变是最为常见的,约占 85%,NRAS 和 HRAS 分别占 12% 和 3%。在美国,死亡率最高的三种癌症(胰腺癌、结直肠癌和肺癌)也恰好是 RAS 突变最多见的三种癌症,分别占这三种癌症患者数的 95%、52% 和 31%。而在乳腺癌、卵巢癌和脑癌中,RAS 的突变非常少见。在胰腺癌、结直肠癌和肺癌中,KRAS 突变占绝对多数。NRAS 突变多见于黑色素瘤和急性骨髓性白血病,HRAS 突变多见于膀胱癌和头颈癌。不过,为什么 KRAS 突变最常见,以及为何不同肿瘤中 RAS 突变发生几率不一样,目前还没有一个很好的解释。
过去 RAS 药物研发过程的经历
三个 RAS 基因编码了 4 个非常相似的蛋白质:KRAS4A、KRAS4B、HRAS 和 NRAS,它们都是三磷酸鸟苷水解酶(GTPases)。RAS 基因的突变会导致其编码的 GTPases 处于持续活跃状态,激活下游信号通路,促进癌细胞生长。
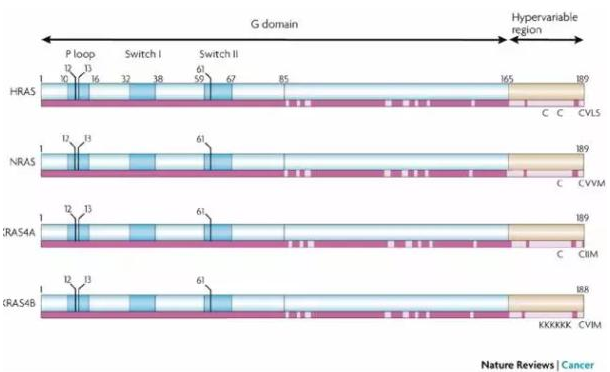
▲RAS 蛋白的四种亚型(图片来源:《Nature Reviews Cancer》)
最早的 RAS 药物研发想法是寻找 GTP 的类似物以抑制 RAS 的活性,不过,由于 RAS 对于 GTP 的亲和度高达皮摩级,而细胞内的 GTP 浓度要高得多,因此这一想法一直没有取得成功。此外,由于对 RAS 与 GTP 结合的活性位点的结构缺乏足够了解,寻找能直接抑制活性部位的药物的努力也失败了。研究人员也尝试过减少 RAS 与细胞膜的结合以抑制其活性。但是,由于当时误以为 4 个 RAS 的功能完全一致,在体外研究中对于 HRAS 有效的药物,在针对 KRAS 占主导地位的癌症的临床试验中也遭到了失败。
近年来,由于对于这四个 RAS 蛋白功能和信号调控的进一步认识,在吸取了以前失败经验的基础上,药物研究又重新活跃了起来。
抑制 RAS 与细胞膜的结合
研究发现,RAS 的致癌功能与其在细胞内的位置有很大关系,当 RAS 位于细胞膜的内表面时活性最强。最近的研究还发现了对 RAS 蛋白翻译后的修饰控制了其在细胞内的位置。在 RAS 转移去细胞膜的过程中的第一步是由法呢基转移酶(farnesyltransferase)在 RAS 的 C 端加上一个法呢基。因此,在 90 年代时,抑制法呢基转移酶的活性是 RAS 药物研发中的重点,不过其中至少两个药物都在 3 期临床中失败了。后来发现,4 个 RAS 蛋白中最常见的 KRAS4B 可以得到一个与法呢基结构类似的香叶基香叶基异戊二烯(geranylgeranyl isoprenoid)来完成向细胞膜的转移。最近,通过筛选可以直接抑制 RAS 与细胞膜结合的小分子,发现了一个潜在的药物 fendiline,它可以选择性地减少 KRAS4B 与细胞膜的结合。
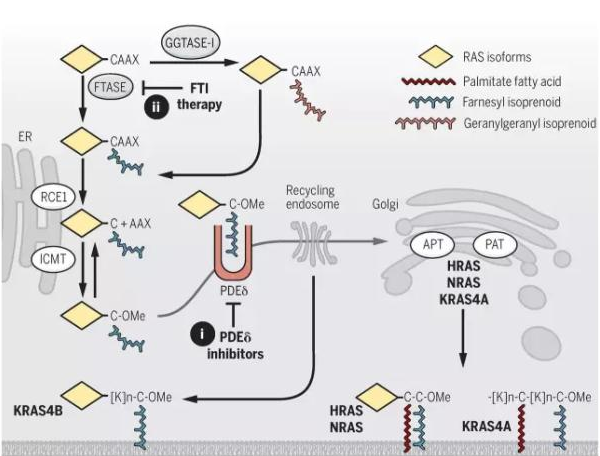
▲RAS 胞内定位和与质膜结合的调控(图片来源:《科学》)
最近的研究中发现的这一通路中另一个潜在的靶点是磷酸二酯酶δ(PDEδ)。PDEδ可以结合 RAS 蛋白上的法呢基,并将 RAS 送往高尔基体,随后在通过囊泡运输前往细胞膜。研究发现了两个小分子药物 deltarasin 和 deltazinone 可以抑制 PDEδ与法呢基的结合。在小鼠肿瘤模型中的实验发现,这两个药物可以将 RAS 滞留在细胞内部从而抑制肿瘤生长。不过这两个药物对于 RAS 不是特异性的,它们有可能影响其它带有法呢基的蛋白的转运,因此其安全性还有待验证。
抑制 RAS 的下游信号通路
抑制 RAS 下游的信号蛋白是目前吸引了最多注意力的针对 RAS 的药物研发策略。不过,RAS 下游已知有至少 11 个不同蛋白家族参与其信号传递,针对哪个或者哪几个家族能产生抗癌效果是一个关键问题。目前研究最集中的是两个信号通路:RAF-MEK-ERK 和 PI3K-AKT-mTOR。因为这两个通路中都被发现有致癌的基因突变,包括 BRAF 和 PIK3CA。目前的研究结果显示,似乎 RAF-MEK-ERK 通路在癌症中更重要一些。因为在没有 KRAS 突变的少量胰腺癌中,一大部分带有 BRAF 突变。在小鼠中的研究显示,如果 RAS 的功能缺失,RAF-MEK-ERK 信号通路中的成员能够弥补这一小点。
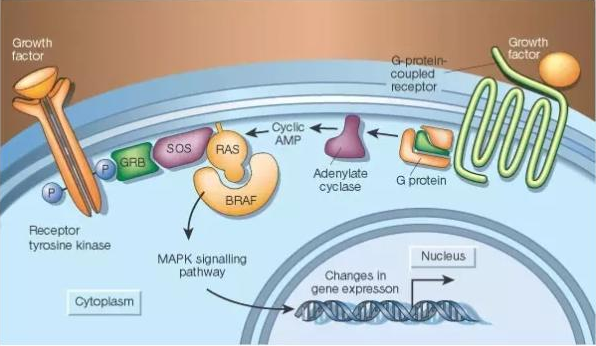
▲RAF-MEK-ERK 通路(图片来源:《自然》)
在 RAF-MEK-ERK 通路中,第一个被 RAS 激活的成员是 RAF 激酶,包括 ARAF、BRAF 和 CRAF。其中,BRAF 抑制剂 vemurafenib 和 dabrafenib 已经获批用于治疗携带 BRAF 突变的黑色素瘤。但是,这两个药物在携带 RAS 突变的肿瘤中不起作用。主要原因是 RAS 突变能够导致 BRAF 和 CRAF 的二聚物形成,抑制 BRAF 反而能够激活 CRAF,从而使 BRAF 抑制剂失去抗癌作用。目前有企业正在研究第二代的泛 RAF 抑制剂,包括 LY3009120 和 PLX8394,它们能够有效抑制下游的 MEK 和 ERK 活性。
MEK 的选择性抑制剂,包括 trametinib 和 cobimetinib 也已经获批用于治疗携带 BRAF 突变的黑色素瘤。不过令人遗憾的是,它们对于 RAS 突变的肿瘤同样不起作用。因为在 RAF-MEK 的信号传导过程中有一个反馈机制,当 MEK 被抑制后,细胞能够加强 RAF 的活性,直到能够克服对于 MEK 的抑制。于是,对于 RAS 突变的药物研发将目光转向了更下游的 ERK 激酶,目前有多个 ERK 抑制剂在临床试验中。

▲PI3K-AKT-mTOR 通路(图片来源:《Nature Reviews Drug Discovery》)
PI3K-AKT-mTOR 通路在肿瘤生长的过程中的作用也许是小了一些,但是它对于 RAF-MEK-ERK 通路有重要的补充作用。对于 RAF-MEK-ERK 抑制剂的抗药性有很大程度就来自于 PI3K-AKT-mTOR 通路的激活。多个药物联用来同时抑制这两个通路在小鼠实验中取得了很好的抗癌效果。但是,在人的临床试验中,同时抑制两个信号通路带来了许多副作用。目前正在研究如何协调对这两个通路的抑制来降低毒性。
RAS 突变的合成致死相互作用
合成致死的概念也被应用到了针对 RAS 的药物研发中。合成致死是指通过大规模筛选寻找那些对于 RAS 突变的细胞而不是 RAS 正常的细胞的存活起关键作用的基因,当抑制那些基因时,与 RAS 突变产生了合成致死相互作用,对正常细胞没有影响。2009 年时,有研究团队报道了 STK33 和 TBK1 等几个激酶与 RAS 突变有合成致死相互作用,但是进一步的研究没有能够证实这些作用。传统上的基于 RNAi 的合成致死基因筛选存在着特异性不高等缺陷是导致这一结果的可能原因。
最近,一些科研人员开始采用特异性更好效率更高的 CRISPR 技术开始寻找 RAS 突变的合成致死基因,并取得了一些进展。被发现的基因包括与 RAS 和细胞膜结合有关的 RCE1 和 ICMT,以及能够调控 ERK-MAPK 信号传导的 RAF1,SHOC2 和 PREX1 等。不过,由于每个携带 RAS 突变的肿瘤都可能存在许多其它突变,这些合成致死基因能有多大的效果还需更多研究。
抑制 RAS 调控的新陈代谢过程
癌细胞的一大特点是其新陈代谢与正常细胞有很大的区别,因为癌细胞需要大量的营养和能量来支持其快速的生长过程。最近的一些研究显示 RAS 突变调控了其中一些过程,如果能够抑制这些过程,也有望抑制 RAS 突变肿瘤的生长。
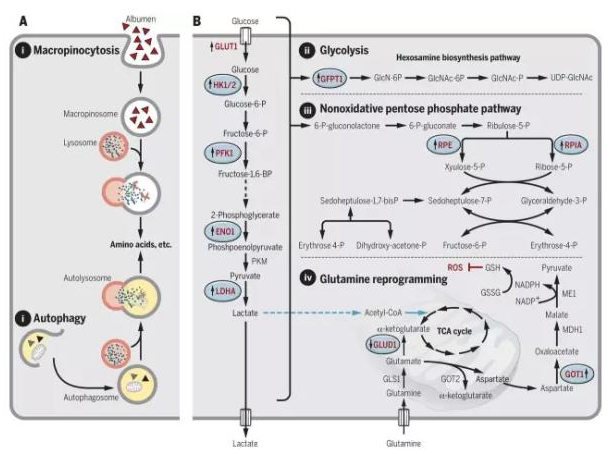
▲RAS 突变带来的癌细胞新陈代谢改变(图片来源:《科学》)
RAS 突变能够激活巨胞饮作用,它是细胞内吞作用的一种,可以从细胞外获取营养物质和水。现在并没有能够直接抑制巨胞饮作用的药物,但是,有一个钠离子 / 氢离子交换通道的抑制剂 EIPA 能够间接抑制巨胞饮作用,有证据显示它能够减缓胰腺癌的生长。另外,巨胞饮作用获取的营养成分中很重要的一种是白蛋白,它可以作为氨基酸的来源。如果使用一些能够与白蛋白特异结合的,携带化疗药物紫杉醇的纳米颗粒,可以将紫杉醇直接送入癌细胞内,也许能取得一定的抗癌效果。
RAS 突变的癌细胞获取营养的另一种方法是自噬作用,细胞可以通过它来降解细胞内不需要的细胞器来获取生长所必需的氨基酸、脂质和核酸等。氯喹类的抗疟药对于自噬作用有抑制效果,并且对于巨胞饮作用也有一定的抑制效果。氯喹在一个小鼠胰腺癌模型中取得了不错的效果。目前有一项临床试验正在评估羟氯喹在胰腺癌中的潜在疗效。
最后,携带 RAS 突变的胰腺癌细胞被发现更依赖于从谷氨酰胺来产生 NADPH,NADPH 对于维持细胞内的氧化还原平衡起关键作用。正常细胞主要从葡萄糖产生 NADPH,因此,抑制从谷氨酰胺产生 NADPH 的化学过程能够特异针对癌细胞。目前,有一个抑制这个过程中的第一步谷氨酰胺酶的小分子药物 CB-839 正在临床试验中,用于评估其对白血病和多种肿瘤的效果。抑制这个过程中的其它参与者的药物也有多个在临床前研究中。
直接抑制 RAS 的活性
当 1989 年 HRAS 的晶体结构首先被报导时,科研人员发现在它的表面没有一个明显的结合 GTP 的“口袋”,这让那些想要直接抑制 RAS 活性的人感到无从下手。近来,通过大规模的化合物筛选,发现了一些有望直接对 RAS 产生作用的化合物。其中一些能够抑制 RAS 与 SOS1 的结合,这能够有效减慢 RAS 获得 GTP 的速度,另一些能够抑制 RAS 与下游信号蛋白如 RAF 的结合。不过,这些化合物与 RAS 的亲和性都较低,要在人体内产生效果,还需要更高亲和性的化合物。
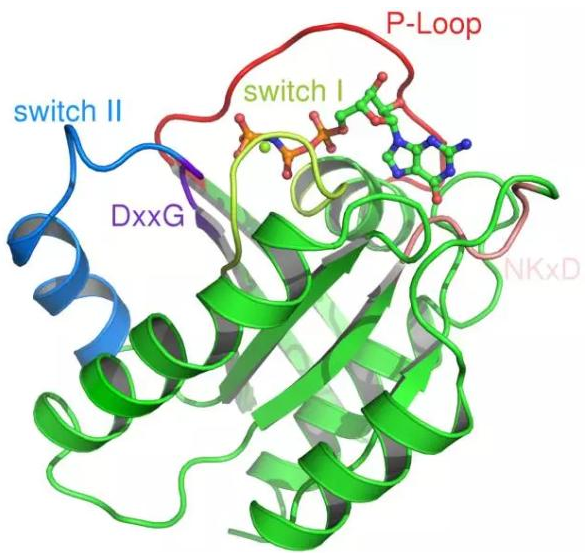
▲RAS 蛋白结构(图片来源:维基百科)
另一些研究人员发现了一类能够抑制 KRAS 的一个特定突变 G12C 的化合物。尽管 G12C 只占所有 KRAS 突变的 12%,它在肺癌中却占大多数。这类化合物能共价结合带 G12C 突变的 KRAS,并将其锁在非激活状态。这其中的一个化合物 ARS-853 在体外实验中能够抑制肺癌细胞的生长。研究还发现,G12C 这个突变和其它突变不同,带 G12C 的 KRAS 的激活过程还与 EGFR 有关。因此,同时抑制 EGFR 和带 G12C 的 KRAS 在理论上能在肺癌中产生更好的疗效。这项研究说明了 KRAS 的不同突变的致癌机制不尽相同,因此,寻找针对特定突变的药物将是下一个研究方向之一。
总结和展望
尽管过去二三十年来对于 RAS 的药物研发过程非常曲折,但是整个领域仍然对前景感到乐观,针对 RAS 的药物在不久的将来应该就会出现。不过我们对于 RAS 的功能以及 RAS 突变致癌的机理仍然没有完全清楚,这意味着在这个领域仍然很有可能出现更多失败的例子。
有一点可以肯定的是,针对 RAS 的药物将不可避免地向个体化发展,出现一个对所有 RAS 突变都有效的药物几乎是不可能的。针对不同肿瘤中的不同 RAS 蛋白,甚至不同的 RAS 突变,都将会有不同的药物。
随着越来越多的科学家加入 RAS 研究的领域,药物研发的过程也将会被加快。从目前来看,针对 RAS 下游信号通路的抑制剂有望最早获得成功,因为这一部分已经有多个药物进入了临床试验。从长远来看,直接抑制 RAS 的药物很有可能是最有效的。随着癌症免疫疗法的迅速发展,RAS 和肿瘤免疫之间的关系会将是一个研究热点。此外,通过 RNAi 或 CRISPR 技术直接抑制 RAS 突变基因的表达也有很大的潜力。
总的来说,尽管仍有许多的障碍,我们对于最终战胜 RAS 突变依然充满了信心。
参考资料:
[1] Drugging RAS: Know the enemy
[2] Stocking oncology's medicine cabinet