昆明动物所开发出适用于高通量异质性数据算法 并成功揭示泛肿瘤存在基因表达紊乱共有模式
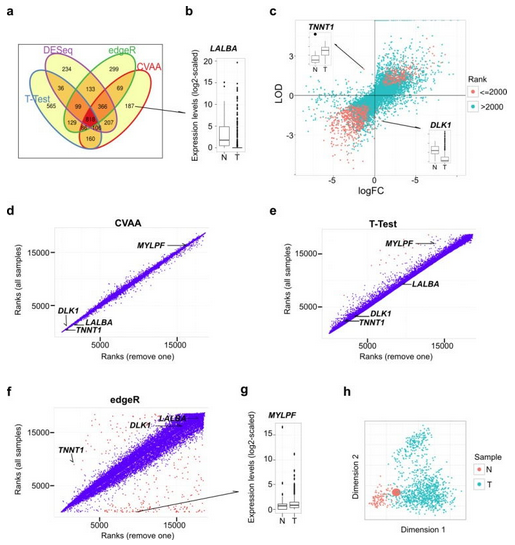
图 1 CVAA 与常用算法的比较
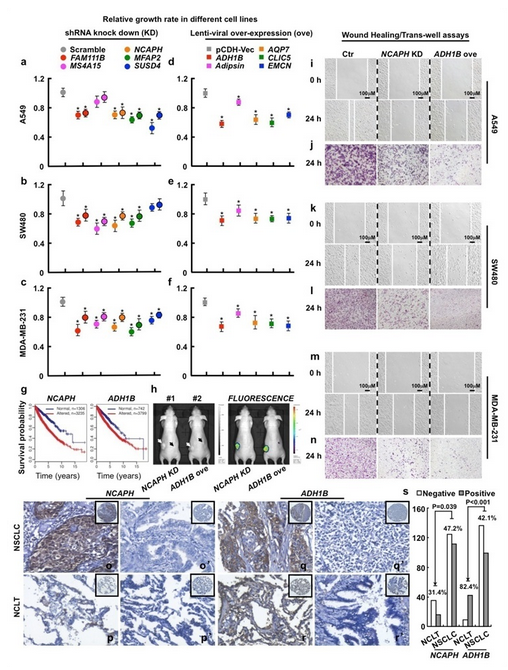
图 2 体内外实验结果
挖掘肿瘤大数据有助于识别和总结肿瘤发生、发展过程的分子变化规律。然而,肿瘤组织高度异质性、批次效应等因素是肿瘤数据分析的重要难题,而目前常用的转录组数据分析方法对于肿瘤离群值极度敏感,容易产生假阴性结果。针对此,中国科学院昆明动物研究所科研人员开发了一种新的不依赖均一化、非参的高维大数据分析算法(Cross-Value Association Analysis,CVAA)。基于对同一批转录组数据(1037 个乳腺肿瘤和 110 个正常组织)的分析结果揭示:与 T -Test、edgeR 及 DESeq 等常用算法相比,CVAA 在处理异质性数据时能明显地减少异常样本的影响和假阴性结果。
进一步将 CVAA 应用于 TCGA 数据库的 5540 个肿瘤(13 种实体瘤)及正常组织的 RNA-Seq 转录组数据,研究人员成功鉴定到大量肿瘤显著差异表达基因,且不同肿瘤之间存在很多相似的基因转录紊乱模式。进而,针对甄别得到的新的肿瘤相关基因和生物学途径,如酒精代谢途径(ADH1B 基因)、补体途径(Adipsin)等,研究人员筛选了 10 个未被报道的基因进行了体内外功能验证研究,结果发现其表达量变化均能显著抑制肿瘤细胞的增殖和 / 或迁移,且部分基因(ADH1B 和 NCAPH)表达量改变显著影响病人生存率和体外移植瘤的生长。因此,该项工作不但成功鉴定出大量新的肿瘤相关基因和通路,为深入理解肿瘤发生发展提供研究靶标,同时也表明 CVAA 算法在大批量、异质性数据分析中具有重要的应用价值。
该研究成果于近期发表在国际期刊 Theranostics 上,昆明动物所助理研究员李其刚、副研究员何永捍、博士生吴焕和副研究员杨翠萍为共同第一作者,研究员孔庆鹏和陈勇彬为共同通讯作者。该项目得到中科院、国家自然科学基金、云南应用基础研究和中科院青年创新促进会等的项目的支持。