胆管癌首个靶向药获批:Pemigatinib为什么不是对所有的FGFR变异都有效?

从FIGHT-202研究来看Pemigatinib 对不同FGFR变异的反应
我们看到FDA基于FIGHT-202研究Cohort A的结果批准Pemigatinb用于携带FGFR2融合或重排的胆管癌的二线治疗。但是其实该研究中还有另外2个Cohort:携带其他FGF/FGFR变异的Cohort B和无任何FGF/FGFR变异的Cohort C。

图 1 FIGHT-202研究设计
从FIGHT-202研究来看,Pemigatinb似乎仅对FGFR2融合/重排的病人有效。对于携带FGFR2融合/重排的Cohort A 客观缓解率(ORR)35.5%,疾病控制率(DCR)达到82.2%, 而Cohort B(n=20)和Cohort C(n=18) 无1例达到客观缓解, DCR分别为40% 和 22.2%,FPS的差异也非常明显(图 2)。几乎可以认为携带其他FGF/FGFR变异的病人与无变异组没有差异。为什么仅有FGFR2融合/重排人群有效呢?回答这个问题之前需要先了解FGFR激活的机理。
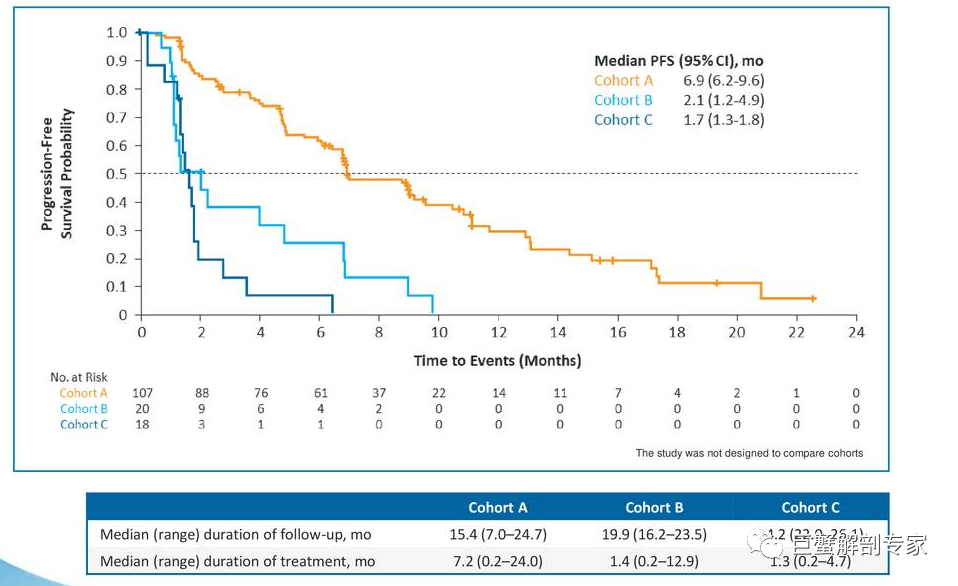
图 2 3个Cohort的PFS数据
关于成纤维细胞生长因子受体(FGFR)结构和激活模式
FGFR属于酪氨酸激酶受体(RTK)大家族,因其在细胞生长/增殖/分化中的重要作用,其变异在肿瘤中十分普遍。FGFR家族有4个成员,FGFR1-4,它们都有着相同的结构,即与配体结合的胞外区(ECD),跨膜区(TMD),近膜区(JMD)和1个“裂开”的激酶区(KD)(图3 ,左)。通常,在配体和肝素的诱导下,FGFR形成二聚体,导致激酶区的“反式”自磷酸化,进而激活下游信号通路(图3 中)。另外一种激活模式针对的是已经形成二聚体的FGFR,配体和肝素分子直接诱导构象发生变化,引起“反式”磷酸化激活(图3, 右)。
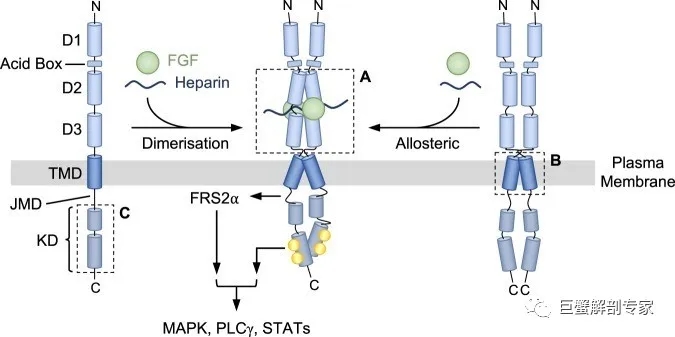
图3 FGFR的激活模式
FGFR2激酶区是如何激活的
早在2001年FGFR2的蛋白质结构已经被解析出来,之后陆陆续续的蛋白质结构学上的工作逐步揭开了FGFR2激酶区激活过程的细节。在非激活状态下,激酶区处于自我抑制状态,激酶区中的一段被称为”刹车”区域的肽段,遮挡住了作为底物的磷酸化位点。在FGFR2非激活状态下,544位的精氨酸(H544)与549位的天冬氨酸(N549), 565位的谷氨酸(E565)与641位的赖氨酸(K641)形成的两道氢键将激酶区紧密地“聚拢”在一起(图4 A 浅灰色),而激活状态下,这2道氢键被打开,激酶“刹车区”被释放开来(图4A 蓝紫色),这一变化进而带动整个激酶区从自我抑制的封闭状态 “伸展”开来,暴露出磷酸化位点。形成“刹车”氢键的几个氨基酸因为其在自我抑制中的重要作用,也是肿瘤中突变发生的热门位点。
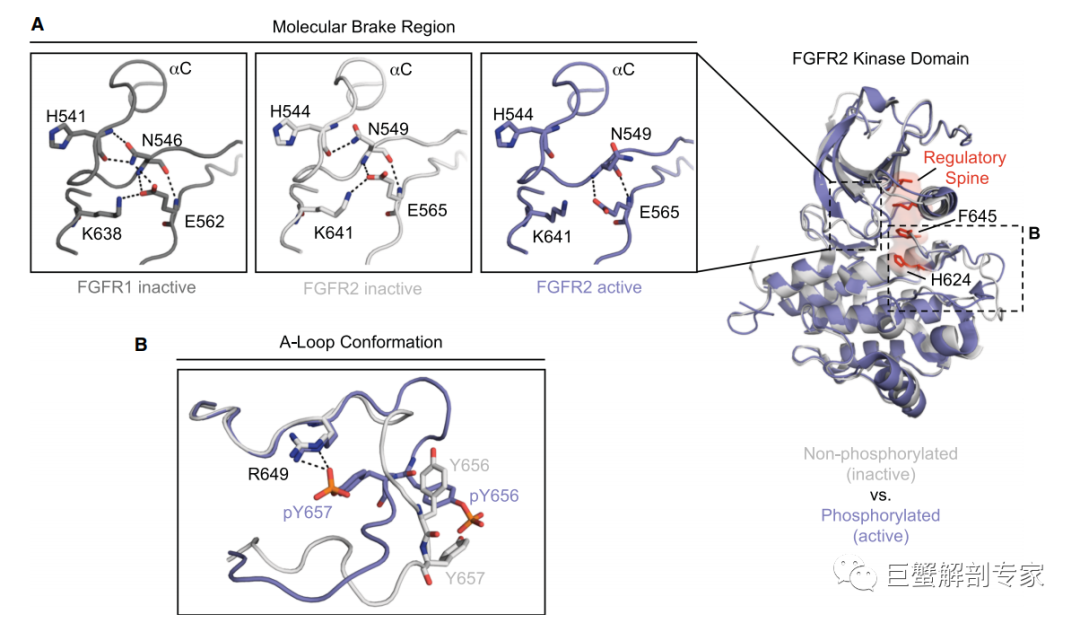
图4 FGFR2 激酶区的激活
从变异类型角度进行分析
FGFR通路常见的变异类型有FGF/FGFR的扩增或过表达,FGFR点突变,FGFR融合/重排。FGFR点突变按发生部位通常又分为胞外区突变,这类点突变可以促进二聚体形成或增强与配体的结合能力;激酶区突变,这类点突变可以不依赖配体直接增强激酶活性;融合,融合也分为I型和II型,FGFR位于3’端的为I型,位于5’端的为II型, FGFR2的绝大多数融合属于II型。
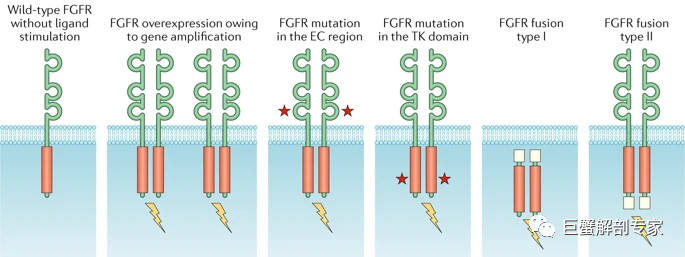
图 5 FGFR的变异类型
既是激活变异也是主要驱动突变的FGFR变异才可能从FGFR抑制剂治疗中取到较好的效果。对于FGFR2融合。5’端的FGFR区域通常保留了完整的激酶区,同时3’端融合伴侣的存在破坏了FGFR激酶区的“封闭式”结构且会诱导FGFR二聚体的形成导致不依赖配体的持续性激活。FGFR2的融合与胆管癌其他主要驱动突变如KRAS, IDH1,可靶向激酶变异互斥出现,也表明其是主要驱动突变之一,同时满足了两个条件。Pemigatinib在FGFR2融合的胆管癌中也确实取得了很不错的疗效。
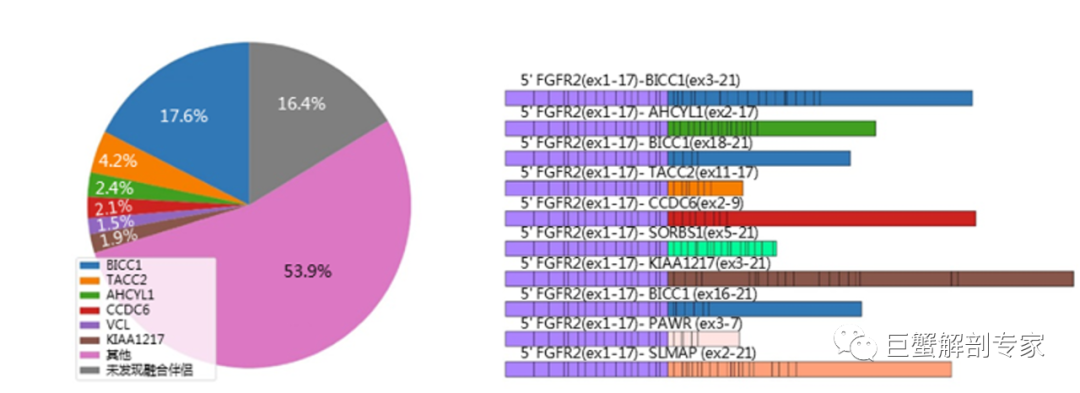
图 6 FGFR2的融合伴侣分布及常见的10种融合
对于FGF/FGFR扩增或者过表达。FGF扩增/过表达增加配体的数量,FGFR的扩增/过表达增加了受体形成了二聚体的几率,对下游通路也有的激活效果,尽管这两种变异对下游通路的激活是否如FGFR融合那样强力还有待研究。但是,携带FGF/FGFR扩增或过表达在胆管癌中通常还有其他强力驱动突变,如HER2,KRAS,此时,FGF/FGFR扩增往往不是肿瘤发生发展的主要矛盾,给予这类病人FGFR抑制剂单药治疗效果往往不尽如人意。
对于FGFR点突变,情况会更复杂一些。激活突变的作用机制可以归为两类,一类是增强激活构象的稳定性,另一类是解除激酶区的自我抑制状态。胞外区的突变通常是通过影响二聚体形成或者增强与配体亲和力激活下游通路。以FGFR3举例(点突变主要发生于FGFR3, FGFR1/2也有类似突变,位置略有不同),其R248C/S249C突变,这两处位点的突变会导致不依赖配体的二聚体形成,即增强了激活构象的稳定性。有类似效果的还有跨膜区的G375C/G370C。激酶区的突变通常直接改变FGFR激酶活性。如位于“刹车区”的N540X突变或者模拟磷酸化状态的K650E突变。这些突变都是经典的激活突变并且已经在临床中得到验证,如Erdafitinib对携带FGFR2/3激活突变的膀胱癌有不错的疗效。对于胆管癌来说FGFR3的变异比较少见,且研究表明在实体瘤中存在着大量的不影响激酶活性的FGFR中性变异,显然携带这些变异的患者很难从FGFR抑制剂中获益。因此,对于点突变具体突变要具体分析。
考量靶向药物对于特定靶点的有效性,不仅要看变异是否影响通路功能,还要看变异是否为主要驱动变异。对于FIGHT-202研究Cohort B,研究没有公布这20例病人具体的变异形式,我们不知道有多少是非FGFR2的融合,多少是点突变(什么位点)或者扩增(扩增倍数),但从胆管癌的基因突变图谱和临床效果推测,绝大多数应属于FGF/FGFR的扩增。
从药物的角度分析
根据与FGFR结合位置的差异可以分为type I 和Type II,根据与FGFR结合是否可逆可分为可逆性和不可逆性抑制剂。不可逆性抑制剂可以设计成特异性靶向一种FGFR亚型,因此通常FGFR4抑制剂都是不可逆性抑制剂。Pemigatinb属于I型可逆性FGFR抑制剂,对FGFR1-3均有极好结合活性。因此理论上Pemigatinb对于FGFR1-3的各种激活变异都应有不错的抑制效果,但是不同的FGFR抑制剂由于其与FGFR结合方式和结合位点的差异,它们的选择性,活性以及耐药突变也是不同的。例如,在胆管癌中Pemigatinb对FGFR2融合人群有效;而在膀胱癌中Erdafitinib似乎对FGFR3点突变人群效果更佳,对FGFR2/3的融合效果较差。这其中药物本身的差异,不同癌种基因背景的差异可能都有影响,尚需要更多探索。
结语:
越来越多筛选分子靶点的靶向药物获批,意味着肿瘤的治疗越来越精细化。但同时也意味着对肿瘤医生的要求越来越高。我们看到一些不区分瘤种只基于特定biomarker的药物获批,但同时也看到某一瘤种有效的靶向药物并不都能平推到携带同一biomarker的其他癌种。癌症如同一个巨大的迷宫,我们目前是从不同的方向拎起一条条不同的线索向前摸索,时而遇到惊喜,时而撞得头破血流。不知道是否存在一种方法可以鸟瞰迷宫全貌,从而制定出最佳的前进路线。不管怎样,胆管癌迎来了第一款靶向药,值得高兴!
参考文献:
1. https://www.fda.gov/news-events/press-announcements/fda-approves-first-targeted-treatment-patients-cholangiocarcinoma-cancer-bile-ducts
2. Annals of Oncology (2019) 30 (suppl_5): v851-v934. 10.1093/annonc/mdz394
3. https://dailymed.nlm.nih.gov/dailymed/drugInfo.cfm?setid=2a8aa5c0-6c92-4566-8c45-e8f4d1fc20ee
4. Lancet Oncol. 2020 Mar 20. pii: S1470-2045(20)30109-1. doi: 10.1016/S1470-2045(20)30109-1.
5. Cells. 2019 Jun; 8(6): 614. doi: 10.3390/cells8060614
6. Biochemical Society Transactions (2018) 46 1753–1770
https://doi.org/10.1042/BST20180004