首个降解KRAS的PROTAC
2020年4月10日,CREW(Arvinas公司创始人)团队在预印本ChemRxiv发表了“Targeted Degradation of Oncogenic KRASG12C by VHL-recruiting PROTACs”的文章,开发了降解KRASG12C突变体的PROTAC分子LC-2。之前已有利用PROTAC降解内源性KRAS的研究,但未对KRAS进行有效降解。LC-2能快速降解不同纯合体和杂合体肿瘤细胞中的KRASG12C,是报道的首个可以降解内源性KRASG12C的化合物。

摘要:
KRAS在约20%的人类癌症中发生了突变,尽管历史上一直被认为是“不可治疗的”,但它却是药理学调控中最受欢迎的靶标之一。近年来,有效的KRASG12C突变体共价抑制剂的发现引发了针对KRAS的小分子研究的新潮流。尽管这些抑制剂在临床上已显示出希望,但我们希望探索PROTAC介导的降解,作为调节突变KRAS的补充策略。在此,我们报道了LC-2的发展,LC-2是第一个能够降解内源性KRASG12C的PROTAC。LC-2与MRTX849战斗部共价结合KRASG12C并募集E3连接酶VHL,诱导KRASG12C快速和持续降解,导致纯合和杂合KRASG12C细胞系中的MAPK信号传导受到抑制。LC-2证明PROTAC介导的降解是减弱癌细胞中致癌性KRAS水平和下游信号传导的可行选择。
简介:
KRAS编码与膜结合的小GTP酶,该GTP酶从受体酪氨酸激酶(RTK)传递信号,促进细胞增殖,细胞分化或细胞凋亡。在正常细胞中,KRAS起到分子开关的作用,在无效的GDP结合的“关闭”状态和有效的GTP结合的“开启”状态之间循环。该开关受到鸟嘌呤核苷酸交换因子(GEF)蛋白质的严格调控 ,将GDP交换为GTP和GTPase激活蛋白(GAPs),从而增强KRAS7-9本质上缓慢的GTPase活性。GEF和GAP蛋白在KRAS上两个浅结合口袋中的一个或两个处结合,分别称为开关I(残基30-38)和开关II(残基59-76),其构型在GDP和GTP结合状态之间发生了巨大变化。体细胞KRAS突变减弱了该蛋白的GAP介导的酶活性,导致GTP结合的活性KRAS的积累和下游信号的过度激活,导致不受控制的细胞增殖。鉴于其表面上传统的可药物化口袋的稀缺性,KRAS仍然是一个具有挑战性的治疗目标。
KRAS p.G12C突变在肺腺癌(LUAD)中非常普遍。KRASG12C突变体占所有KRAS突变LUAD肿瘤的50%以上(占LUAD肿瘤总数的13%)。此外,3%的大肠癌和所有其他1%的实体瘤表达KRASG12C。这种突变大大降低了KRAS固有的GTPase活性,从而导致用于GTP结合的活性KRAS的积累。最初由Shokat小组领导的最新进展已经确定了与KRASG12C突变半胱氨酸共价和选择性结合的分子。这些化合物在KRAS switch II区域内诱导出一个新颖的类似药物的口袋。负责结合半胱氨酸的亲电试剂的优化以及药物诱导的囊袋内的分子相互作用导致了口服生物利用型KRASG12C抑制剂的开发.由Wellspring,Amgen和Mirati开发的ARS-1620 / ARS-3248,AMG510和MRTX849 已分别显示在体外和体内有效抑制KRASG12C活性。此外,ARS-3248,AMG510和MRTX849已进入I期临床试验,并显示出令人鼓舞的结果。然而,尽管取得了成功,但已经报道了抑制剂治疗后快速的适应性抗药性和MAPK信号转导激活。因此,互补治疗策略的开发可以帮助实现靶向KRAS突变体治疗癌症的全部潜力。
蛋白水解靶基因嵌合体(PROTAC)已经成为一种新的有前途的模式药物开发。这些双功能分子同时与目的蛋白(POI)和E3连接酶结合,形成三元复合物,使E3连接酶能够在近端赖氨酸残基上泛素化POI。随后,泛素化的POI被26S蛋白酶体识别并降解。靶标降解的主要优势是消除了传统的小分子抑制剂通常不会减弱的支架作用。Graygroup最近发布了结合ARS-1620和cereblon E3连接酶配体沙利度胺的PROTAC。这些分子与KRASG12C结合并降解人工GFP-KRASG12C融合蛋白,但无法降解内源性KRAS。本文中,我们报道了一流的内源性KRASG12C降解剂LC-2的开发,该降解剂结合了MRTX849和VHL E3ligase配体。我们观察到纯合和杂合表达KRASG12C的细胞中通过真正的PROTAC机制的快速降解。在多种癌细胞系中KRASG12C的急性和持续降解使LC-2成为研究KRAS生物学的有价值的工具化合物,并代表了基于PROTAC的候选疗法发展的重要一步,该疗法通过诱导致癌性KRAS降解而发挥作用。
结果:
1.基于MRTX849的VHL招聘PROTAC参与并降解内源性KRASG12C非纯合和杂合突变细胞系。
鉴于其有希望的I期临床数据和综合的可延展性,我们选择MRTX849(MRTX;图1A)作为设计靶向KRAS的PROTAC的起点。将MRTX对接到KRASG12C的“ switch II”口袋中,可以发现吡咯烷基被溶剂暴露(PDB:5V9U;SI图1A)。为了避免在吡咯烷的2,3或4位引入另一个立体中心并使我们的合成路线进一步复杂化,我们决定从吡咯烷的N-甲基部分构建接头。我们看到了KRAS与LC-1结合的第一个证据(图1A和B)。当以浓度不断增加的LC-1处理NCI-H2030细胞24小时时,我们在1,2.5、10和25μM处观察到了明显的带移 ,表明存在PROTAC偶联的KRAS(图1B)。但是,仅观察到KRAS含量略有下降,但没有显着降低。因此,这些数据表明LC-1可以参与KRASG12C,但不能有效降解该蛋白质。结果,在我们的PROTAC筛选中,LC-1随后被用作KRAS参与的阳性对照。

图一:MRTX849-VHL PROTAC参与并降解NCI-H2030细胞中的内源性KRASG12C:A)MRTX849,LC-1(惰性PROTAC),LC-2(活性PROTAC)和LC-2 Epimer的化学结构。B)LC-1以剂量依赖性方式参与KRASG12C。右边的定量。C)LC-2以剂量依赖性方式降解KRASG12C。右边的定量。定量数据代表平均值±SD。不重要(N.S.); * p <0.05;** p <0.01;**** p <0.001
LC-1的一个主要缺陷是在连接基中存在可水解的酰胺。为了解决这一责任,直接从吡咯烷环氮上扩展了后续PROTAC的连接基。我们筛选了一个PROTAC小型文库,其接头长度比LC-1短和长几个原子,并且从该筛选中,我们确定LC-2是最有效降解KRASG12C的PROTAC(图1A)。LC-2在NCI-H2030细胞中诱导的内源性KRASG12C在2.5μM浓度下最大降解,DMax大于75%,DC50为0.59±0.2μM(图1C)。在10μMLC-2下,观察到KRASG12C条带的分子量与LC-1修饰的KRASG12C分子量相同。这种未降解的较高分子量带的出现表明在高LC-2浓度下开始出现“钩效应”。“钩效应”是PROTAC的标志,在高药物浓度下,具有靶标或带有E3连接酶的非生产性二聚体的形成会超过降解所必需的三元复合物的形成。
已知MRTX对KRASG12C突变体具有比其他KRAS突变体更高的选择性。为了探索LC-2的特异性,在具有杂合KRASG13D突变的HCT 116细胞中检查了KRAS降解。在LC-2浓度高达10μM的情况下,未观察到KRASG13D的参与或降解(SI图1B)。这些数据进一步表明,LC-2选择性参与并降解了突变型KRASG12C蛋白。
此外,我们在5种不同的KRASG12C细胞系中测试了LC-2,并观察到DC50值介于0.25和0.76μM之间,DMax值介于> 75-90%之间(表1)。LC-2可以降解纯合子和杂合子细胞系中的突变KRAS,对MRTX21的敏感性不同。我们观察到NCI-H23细胞中杂合子的降解率> 50%。从理论上讲,由于这些细胞带有一种野生型和一种突变型KRASG12C等位基因,因此,如果表达相等,则可以预期最多降解50%,正如我们在NCI-H358细胞中所见(SI图2A)。但是,在使用KRASG12C特异性siRNA进行的siRNA敲除实验中,NCI-H23细胞几乎完全消失了KRAS,这与我们在LC-241上观察到的降解是一致的。这些数据表明,基于MRTX的VHL募集的PROTAC 可以使多种癌细胞系参与和降解KRASG12C。

表1. LC-2在多种KRAS突变癌细胞系中诱导内源性KRASG12C降解:一组KRASG12C癌细胞系中的PROTAC活性。达到最大降解(Dmax)的50%的DC50。
2.LC-2诱导的KRASG12C降解通过真正的PROTAC机制发生。
VHL配体的羟基脯氨酸部分赋予与E3连接酶的结合,而4-羟基脯氨酸部分的绝对立体化学的转化取消了VHL的结合。因此,我们合成了LC-2 Epimer(图1A),作为一种物理化学匹配的阴性对照分子,无法积累VHL。当NCI-H2030细胞用2.5μMLC-2异构体处理4小时时,仅观察到KRAS参与,而2.5μMLC-2诱导显着降解(〜65%;图2A)。
PROTAC通过促进其泛素化来靶向蛋白质通过蛋白酶体降解,这取决于POI,PROTAC和E3连接酶之间的三元复合物(在本例中为VHL)的形成。由于过量的VHL配体会抑制三元复合物的形成,因此我们在NCI-H2030细胞中进行了竞争实验,该细胞在用2.5μMLC-2处理之前用过量的VHL配体预处理了1小时。LC-2与VHL配体的竞争通过阻止PROTAC与VHL的结合而降低了KRASG12C的水平(图2A)。然而,在LC-2处理中观察到的较高分子量的KRASG12C谱带表明,PROTAC仍然能够与KRASG12C结合。
CUL2类泛素化修饰是VHL适配器蛋白,对于E3连接酶复合物42的正确组装和功能是必要的。为了进一步研究LC-2诱导的KRASG12C降解是否通过真正的PROTAC机制发生,在用2.5μM LC-243-44处理之前,将NCI-H2030细胞用1μM的降酰化抑制剂MLN4924或1μM的蛋白酶体抑制剂epoxomicin处理。这两种抑制剂都能抑制KRASG12C水平,表明LC-2对KRASG12C的降解依赖于蛋白酶体和类泛素化(图2A)。
KRAS被束缚在质膜上,KRASG12C的单泛素化可能通过溶酶体途径诱导KRASG12C的内吞和降解.因此,我们还测试了溶酶体酸化抑制剂bafilomycin A1(BafA1)是否可以挽救KRASG12C降解。用BafA1预处理NCI-H23不能挽救LC-2诱导的KRASG12C降解,而烯丙基化抑制作用再次挽救了KRAS降解(图2B)。综合这些数据表明,LC-2诱导的KRASG12C降解取决于与VHL和功能泛素蛋白酶体系统形成的三元复合物,而不取决于溶酶体。

图2:通过PROTAC机制降解内源性KRASG12C。A)LC-2 Epimer在2.5μM时不会诱导KRASG12C降解,并且在NCI-H2030细胞中,VHL配体竞争,环氧化物霉素(Epox)的蛋白酶体抑制和MLN4924(MLN)的酰化抑制可挽救LC-2诱导的降解。定量如下。B)抑制烯丙基化,但不抑制溶酶体酸化,可挽救LC-2诱导的NCI-H23细胞中KRASG12C降解。定量如下。定量数据代表平均值±SD。不重要(N.S.); *** p <0.005
3.LC-2在多种癌细胞系中诱导快速持续的KRASG12C降解
为了探索PROTAC诱导的KRASG12C降解动力学,使用2.5μMLC-2作为固定浓度在NCI-H2030细胞和SW1573细胞中进行了时程实验,因为它在24小时内在所有细胞系中诱导了最大降解(图1A和SI图2)为了区分目标结合率和降解率,LC-2 Epimer被用作监测KRASG12C结合的阴性对照。通过仅将LC-2 Epimer修饰条带的强度与DMSO处理的样品中未结合的KRAS的强度进行比较,就可以实现对结合的定量分析。对于NCI-H2030细胞,两者的KRASG12C结合均早于1小时LC-2和LC-2 Epimer(图3A)。在4小时内发生了最大程度的接触和明显的降解。NCI-H2030细胞在8小时内达到最大降解,并持续长达24小时。SW1573细胞显示更快的动力学,在1小时内接近最大参与度。但是,降解速率比NCI-H2030细胞要慢,因为直到12小时才观察到最大降解(图3B)。
在我们的PROTAC筛选中,我们观察到0.1μMMRTX和10μMLC-1会增加KRAS蛋白水平(图1C)。尽管Hallin等人未观察到MRTX增加KRASG12C蛋白水平,但我们的数据与先前使用KRASG12C抑制剂ARS162019所观察到的一致。因此,我们探讨了用LC-2进行更长的治疗会影响KRASG12C水平的情况。用2.5μMLC-2处理MIA PaCa-2,NCI-H23和SW1573细胞6、24、48和72小时。在所有三种细胞系中,最大的KRAS降解在24小时内发生,并持续长达72小时(图4A-B和图3S)。LC-2 异构体完全参与了SW1573细胞中的KRASG12C,但并未像预期的那样降低蛋白质水平(SI,图3)。在NCI-H23细胞中,KRASG12C在72小时开始反弹。这些数据加在一起表明,LC-2能够在纯合和杂合细胞系中快速且持续地降解KRASG12C。克服增加的KRASG12C表达的能力表明,降解可能比抑制BRK4降解子对延长下游信号的抑制作用更有益。

图3:KRASG12C降解迅速,最大降解最早可在4小时内诱导:A)NCI-H2030细胞的时间进程。LC-2和LC-2差向异构体在1小时内结合,在8小时内观察到最大程度的降解,并维持长达24小时。右边的定量。B)SW1573细胞中的时间进程。LC-2和LC-2差向异构体在1小时内与KRAS结合,在12小时内观察到最大降解,并维持长达24小时。右边的定量。LC-2 Epimer是对更高分子量的PROTAC Epimer修饰条带的定量,可监测KRASG12C随时间的推移而不是总KRAS含量。定量数据代表平均值±SD。不重要(N.S.); * p <0.05;** p <0.01;*** p <0.005; **** p <0.001。

图4:内源性KRASG12C在多种癌细胞系中持续降解超过72小时。A)MIA PaCa-2细胞中72小时的时间过程。降解发生在6小时,并保持长达72小时。右边的定量。B)在NCI-H23细胞中72小时的时间进程。降解在6小时内发生,在24小时达到最大值,并在72小时后开始反弹。右边的定量。定量数据代表平均值±SD。不重要(N.S.); ** p <0.01;*** p <0.005; **** p <0.001
4.LC-2诱导的KRASG12C降解调节纯合和杂合KRAS突变细胞系中的Erk信号传导
在24小时的剂量反应过程中,在NCI-H2030和NCI-H23细胞中研究了LC-2调节Erk信号传导的能力。在NCI-H2030细胞中,检测到pErk,并观察到信号传导的剂量依赖性降低(图5A)。另外,总Erk水平以剂量依赖性方式升高。NCI-H23细胞在pErk中显示出类似的剂量依赖性下降(图5B)。另外,总Erk水平以剂量依赖性方式升高。
在经过2.5μMLC-2处理的MIA PaCa2,NCI-H23和SW1573细胞中,在24小时内监测信号动力学。在MIA PaCa-2和NCI-H23细胞中,MRTX和LC-2对Erk信号的调节均在6小时之内发生(图6A-B和SI图4)。在每种细胞系中,第6和24小时两种化合物均抑制pErk。在SW1573细胞中,磷酸化Erk在1至4小时之间被2.5μMLC-2抑制,但是pErk水平在8至24小时之间回弹(SI图4)。尽管如此,在24小时时,LC-2处理的细胞中的pErk水平仍显着低于DMSO处理的细胞。在所有时间点,与DMSO相比,LC-2处理的细胞中的总Erk均增加,表明在KRASG12C降解和pErk抑制后启动了正反馈环。综上所述,这些数据表明,LC-2诱导的KRASG12C降解能够调节下游信号传导,抑制和降解之间的信号传导差异取决于细胞系。
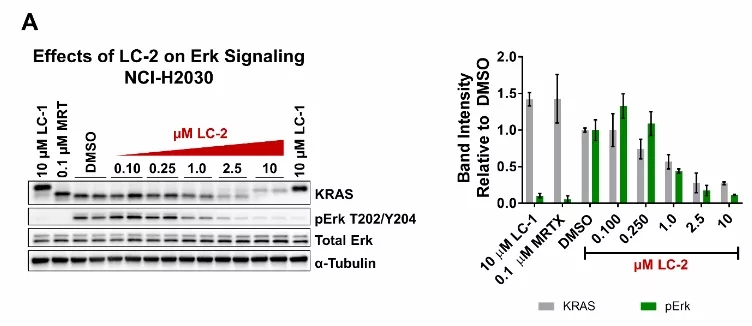

图5:内源性KRASG12C的降解调节了纯合和杂合KRASG12C细胞系中的Erk信号传导。A)在纯合NCI-H2030细胞中KRASG12C的降解以剂量依赖性方式减弱pErk。右边的定量。B)杂合的NCI-H23细胞中KRASG12C的降解以剂量依赖性方式降低了pErk。右边的定量。量化数据表示平均值±SD。
讨论:
