首个降解KRAS的PROTAC
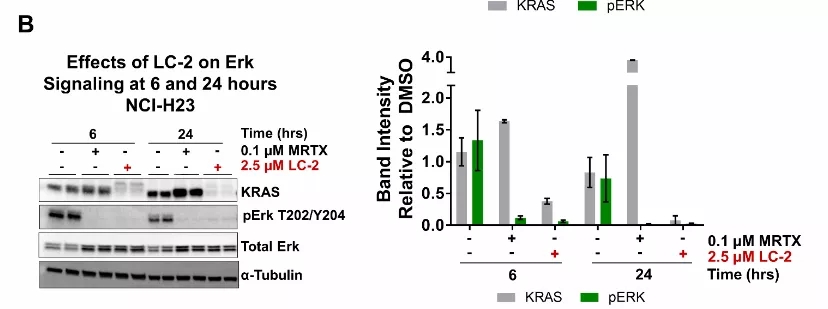
图6:随着时间的推移,KRASG12C降解和抑制对Erk信号的影响。A)在纯合的MIA PaCa-2细胞中,KRASG12C的抑制和降解会在6和24小时降低pErk信号传导。右边的定量。B)在杂合的NCI-H23中,KRASG12C的抑制和降解会在6和24小时降低pErk信号传导。右边的定量。量化数据表示平均值±SD。
据我们所知,这项研究是PROTAC诱导癌细胞内源性KRASG12C降解的首次报道。我们的PROTAC LC-2将共价KRASG12C抑制剂MRTX与我们实验室中开发的VHL配体偶联. VHL募集到KRASG12C诱导了内源性KRAS泛素化和降解,DC50值为0.25至0.76μM。我们观察到快速参与,持续的KRAS降解,并在几种KRASG12C突变细胞系中长达72小时的pErk信号减弱。该工具化合物将有助于进一步探索KRAS降解如何影响下游信号传导和KRASG12C突变型癌细胞的生存力,其时间控制要比基于核酸的敲除方法更为精确。
这项工作不是降低KRASG12C的首次尝试。最近,Zeng等人在24小时内未能成功降解具有20μMXY-4-88的内源性KRASG12C。我们观察到,PROTAC的两个组成配体之间的差异都可能显着影响靶标参与的效率和选择性。进一步的研究将集中于了解KRASG12C配体,募集的E3连接酶或这两种因素在赋予LC-2中的组合的重要性。通过SPR进行三元复合测定和/或通过使用串联泛素结合实体(TUBE)下拉并随后进行免疫印迹监测这些化合物诱导泛素化的能力可以解决这些问题。
随着几种新的共价抑制剂的出现,响应于KRASG12C抑制而进行的激酶组重新布线一直是一个活跃的研究领域。已经发现,MRTX,AMG510和ARS1620衰减的信号在24至72小时之间恢复到或超过基础水平。这与几种酪氨酸激酶的活性增加有关。为了对抗这种获得性抗性,已经成功地使用了FGFR抑制剂的靶向抑制以及SHP2抑制剂的泛RTK抑制与KRASG12C抑制的组合来减少被抑制信号的回收。与单独的RTK抑制或KRASG12C抑制相比,这些共同治疗方案在体外和体内也显示出更抗增殖的作用。确定单独的降解是否可以克服Erk信号再激活和/或KRAS降解与RTK抑制的组合是否可以进一步增强抗增殖作用将是很有趣的。除了重新布线敏感细胞外,还有一些已知的细胞系,例如SW1573(用于本研究)和NCI-H1792,它们固有地对KRASG12C抑制的抗增殖作用具有抵抗力。最近,研究表明在这些细胞中siRNA介导的敲低,但未抑制KRASG12C,导致细胞活力降低了约50%。因此,确定由LC-2引起的KRASG12C诱导的KRASG12C降解在这些细胞系中是否也具有相似的抗增殖性将是有趣的。
用共价抑制剂靶向KRAS的能力本身是药物发现的一个里程碑。结果表明,被正式认为是不可药物的蛋白质可以被小分子靶向。类似地,此处给出的结果表明,只要鉴定出合适的配体,内源性KRASG12C即可被降解。尽管其他KRAS突变体的配体发展仍在继续,但LC-2可以作为一种工具化合物,用于在KRASG12C快速降解(即细胞活力)的背景下研究生物学。我们的前导化合物LC-2的一个警告是PROTAC的共价性质可能会限制它的效力,因为它不能参与降解的催化循环。因此,有必要努力开发针对KRAS突变体的可逆PROTAC。尽管有局限性,但LC-2的发现为在癌症治疗中靶向KRAS突变体提供了新的机会。